Methods and Materials

Before the PLA2s could be analyzed and sequenced, several steps had to be taken. This page gives a general overview of the methods used during this project.
RNA Purification
The samples from the snakes had many components besides PLA2 RNA, and because of this, RNA had to be purified from the samples. RNA was purified from the samples using two different methods. For C. s. tzabcan, the TRIzol kit from Life Technologies was used. For C. basiliscus, the Direct-zol MiniPrep Kit from Zymo Research was used. The RNA was also selected for using oligo(dT) primers from Life Technologies, because these primers attached to the poly-A tails (long strings of A nucleotides) found at the end of the RNA. After being purified, a Nanodrop was used to determine the amount of RNA and its purity in order to ensure that it was clean enough to be converted into cDNA. Nanodrops use wavelengths to determine the concentrations of different molecules because these molecules emit and absorb different wavelengths of light. Specifically, the 260/280 ratio was examined to check the protein contamination and the 260/230 ratio was examined to check for other contaminants such as ethanol. All of the processes with RNA were performed using gloves and tools which had been washed with RNase inhibitor. The processes were also performed in a laminar flow hood which was washed with RNase inhibitor.
RNA Purification
The samples from the snakes had many components besides PLA2 RNA, and because of this, RNA had to be purified from the samples. RNA was purified from the samples using two different methods. For C. s. tzabcan, the TRIzol kit from Life Technologies was used. For C. basiliscus, the Direct-zol MiniPrep Kit from Zymo Research was used. The RNA was also selected for using oligo(dT) primers from Life Technologies, because these primers attached to the poly-A tails (long strings of A nucleotides) found at the end of the RNA. After being purified, a Nanodrop was used to determine the amount of RNA and its purity in order to ensure that it was clean enough to be converted into cDNA. Nanodrops use wavelengths to determine the concentrations of different molecules because these molecules emit and absorb different wavelengths of light. Specifically, the 260/280 ratio was examined to check the protein contamination and the 260/230 ratio was examined to check for other contaminants such as ethanol. All of the processes with RNA were performed using gloves and tools which had been washed with RNase inhibitor. The processes were also performed in a laminar flow hood which was washed with RNase inhibitor.

cDNA Synthesis
The RNA was then synthesized into cDNA (complementary DNA, so named becasue it is only the part of the DNA which codes for the protein).and amplified using touchdown PCR (polymerase chain reaction). The 3’RACE System for Rapid Amplification of cDNA Ends kit from Invitrogen was used for this process. A sense primer with conserved signal peptide regions from PLA2s was used to select for PLA2 RNA, so that only the desired RNA was synthesized into cDNA and amplified. After using the kit, PCR was used to provide the proper conditions for amplifying the cDNA. The kit added polymerase, the enzyme which copies DNA, and free nucleotides to the mixture. By cycling the mixture through a series of temperatures (touchdown PCR), the polymerase was stimulated to copy the RNA and make it into cDNA, and to make many copies of the DNA (amplification).
PCR Gel Electrophoresis
After the cDNA was amplified, a gel electrophoresis was used to check for PLA2 cDNA and to isolate that cDNA. An agarose gel was placed in a running box with the lanes on the side of the negative anode, and buffer over it. The PCR samples were mixed with loading dye, so that the samples could be seen while they were being added to the gel.. A ladder with components of known sizes was also prepared to serve as a comparison to the samples. Once the ladder and PCR samples were ready, they were loaded into the gel, with one sample in each lane. The running box was then connected to a power source and allowed to run for 50-60 minutes at 100 volts. The gel was then removed from the running box and observed in a florescent box to view the bands of PCR product. All of the bands from the samples which had the expected PLA2 transcript size (500-600 base pairs) were removed from the gel using a scalpel.
The RNA was then synthesized into cDNA (complementary DNA, so named becasue it is only the part of the DNA which codes for the protein).and amplified using touchdown PCR (polymerase chain reaction). The 3’RACE System for Rapid Amplification of cDNA Ends kit from Invitrogen was used for this process. A sense primer with conserved signal peptide regions from PLA2s was used to select for PLA2 RNA, so that only the desired RNA was synthesized into cDNA and amplified. After using the kit, PCR was used to provide the proper conditions for amplifying the cDNA. The kit added polymerase, the enzyme which copies DNA, and free nucleotides to the mixture. By cycling the mixture through a series of temperatures (touchdown PCR), the polymerase was stimulated to copy the RNA and make it into cDNA, and to make many copies of the DNA (amplification).
PCR Gel Electrophoresis
After the cDNA was amplified, a gel electrophoresis was used to check for PLA2 cDNA and to isolate that cDNA. An agarose gel was placed in a running box with the lanes on the side of the negative anode, and buffer over it. The PCR samples were mixed with loading dye, so that the samples could be seen while they were being added to the gel.. A ladder with components of known sizes was also prepared to serve as a comparison to the samples. Once the ladder and PCR samples were ready, they were loaded into the gel, with one sample in each lane. The running box was then connected to a power source and allowed to run for 50-60 minutes at 100 volts. The gel was then removed from the running box and observed in a florescent box to view the bands of PCR product. All of the bands from the samples which had the expected PLA2 transcript size (500-600 base pairs) were removed from the gel using a scalpel.
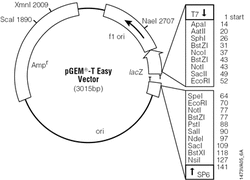
PCR Purification and Insertion into Bacterial Plasmid
The PCR products were removed from the agarose and purified using the Zymoclean Gel DNA Recovery kit from Zymo Research. The PLA2 transcripts were then ligated into a plasmid so that they could be transformed into bacteria to be cloned. A plasmid is a round strand of DNA which can be taken up and used duplicated by bacteria. The ligation was performed using pGEM-T Easy Vector System from Promega. This plasmid contained two genes, ampicillin resistance, which allowed the bacteria which took up the plasmid to live in ampicillin filled environments, and a gene which produced a protein that would turn the bacteria blue when a specific type of sugar was added. The PLA2 transcripts which were inserted into the plasmid interrupted the gene to turn the bacteria blue, which meant that any bacteria which received the PLA2 transcripts would be white in color. This allowed for the selection of bacteria which contained the desired transcripts.
The PCR products were removed from the agarose and purified using the Zymoclean Gel DNA Recovery kit from Zymo Research. The PLA2 transcripts were then ligated into a plasmid so that they could be transformed into bacteria to be cloned. A plasmid is a round strand of DNA which can be taken up and used duplicated by bacteria. The ligation was performed using pGEM-T Easy Vector System from Promega. This plasmid contained two genes, ampicillin resistance, which allowed the bacteria which took up the plasmid to live in ampicillin filled environments, and a gene which produced a protein that would turn the bacteria blue when a specific type of sugar was added. The PLA2 transcripts which were inserted into the plasmid interrupted the gene to turn the bacteria blue, which meant that any bacteria which received the PLA2 transcripts would be white in color. This allowed for the selection of bacteria which contained the desired transcripts.
Cloning
Bacteria were used to clone the transcripts so that there would be enough copies to be sequenced. Specifically, a mutated species of E. coli was used. Because there are many bacteria in the environment, all materials that were used in this part of the experiment had to be sterilized before they ere used. This was done by exposing the materials to intense heat which killed all contaminating bacteria. Agar was poured for the bacteria to live on which contained ampicillin and the sugar which would turn the bacteria blue. The E. coli used were made competent, meaning that they were made extremely fragile o that they would accept plasmids from the surronding environment, then exposed to the plasmids. The E. coli were then added to the agar plates and left to grow so that they would replicate the PLA2 transcripts many times.
Preparation for Sequencing
White bacteria colonies were picked from several locations off of the agar and left to grow more bacteria. The PLA2 plasmids were purified from the E. coli using the Quick Clean 5M Miniprep kit from Genscript Each purified plasmid was then Nanodropped to determine the concentration of the plasmids in nanograms per microliter. For each plasmid sample, it was calculated how many microliters would hold 200 ng of plasmid becasue DNASU required that only 200 ng of each sample be sent,. The appropriate amount of each plasmid was separated out and added to new tubes. These tubes were then sent to the DNASU Sequencing Core to be sequenced using Sanger sequencing. Snager sequncing uses polymerase, gel electrophoresis, and fluouresct dye and lasers to determine the DNA sequences of transcripts.
Bacteria were used to clone the transcripts so that there would be enough copies to be sequenced. Specifically, a mutated species of E. coli was used. Because there are many bacteria in the environment, all materials that were used in this part of the experiment had to be sterilized before they ere used. This was done by exposing the materials to intense heat which killed all contaminating bacteria. Agar was poured for the bacteria to live on which contained ampicillin and the sugar which would turn the bacteria blue. The E. coli used were made competent, meaning that they were made extremely fragile o that they would accept plasmids from the surronding environment, then exposed to the plasmids. The E. coli were then added to the agar plates and left to grow so that they would replicate the PLA2 transcripts many times.
Preparation for Sequencing
White bacteria colonies were picked from several locations off of the agar and left to grow more bacteria. The PLA2 plasmids were purified from the E. coli using the Quick Clean 5M Miniprep kit from Genscript Each purified plasmid was then Nanodropped to determine the concentration of the plasmids in nanograms per microliter. For each plasmid sample, it was calculated how many microliters would hold 200 ng of plasmid becasue DNASU required that only 200 ng of each sample be sent,. The appropriate amount of each plasmid was separated out and added to new tubes. These tubes were then sent to the DNASU Sequencing Core to be sequenced using Sanger sequencing. Snager sequncing uses polymerase, gel electrophoresis, and fluouresct dye and lasers to determine the DNA sequences of transcripts.
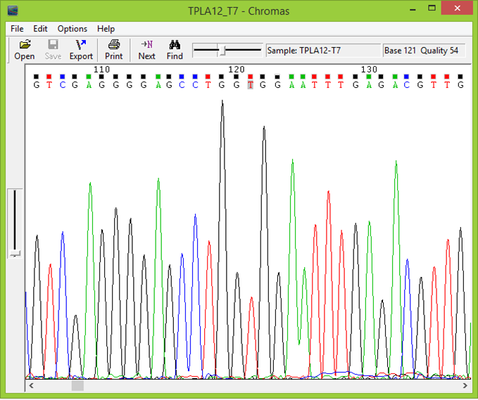
Analysis of Transcripts
Once the PLA2 plasmid sequences returned from the sequencing lab the sequences were examined using Chromas Lite from Technelysium. The sequences were analyzed for potential mistakes and to make sure they were of a high enough quality to be used. The primer nucleotides were then removed from the nucleotide sequences, and the new sequences were translated into amino acid sequences using ExPASy Translate from the Swiss Institute of Bioinformatics. The translated amino acid sequences were then run through BLASt from the National Center for Biotechnical Information in order to compare them to known PLA2s. BLAST listed which proteins the sequences found in this study most resembled, and this information was used to organize the PLA2 sequences for a sequence alignment. Sequences which were found to most closely resemble crotoxin A were run in a sequence alignment with crotoxin A from C. d. terrificus and sistruxin A from Sistrurus catenatus tergeminus, a protein with a similar function to crotoxin A. Sequences which were found to most closely resemble crotoxin B were run in a sequence alignment with crotoxin B from C. d. terrificus, sistruxin B from S. c. tergeminus, and N6 PLA2s from Bothriechis schlegelii and Crotalus viridis viridis. The sequence alignments were made using ClustalW2 from the European Bioinformatics Institute.
Protein Analysis
A protein gel was run using crude venom from both snake species. This gel was run using a 1D 12% polyacrylamide Bis-Tris NuPAGE gel from Life Technologies.The purpose of this gel was to analyze the protein contents of both venoms in order to ensure that they really did contain PLA2 venoms which would be produced by the transcripts isolated in this study.
Once the PLA2 plasmid sequences returned from the sequencing lab the sequences were examined using Chromas Lite from Technelysium. The sequences were analyzed for potential mistakes and to make sure they were of a high enough quality to be used. The primer nucleotides were then removed from the nucleotide sequences, and the new sequences were translated into amino acid sequences using ExPASy Translate from the Swiss Institute of Bioinformatics. The translated amino acid sequences were then run through BLASt from the National Center for Biotechnical Information in order to compare them to known PLA2s. BLAST listed which proteins the sequences found in this study most resembled, and this information was used to organize the PLA2 sequences for a sequence alignment. Sequences which were found to most closely resemble crotoxin A were run in a sequence alignment with crotoxin A from C. d. terrificus and sistruxin A from Sistrurus catenatus tergeminus, a protein with a similar function to crotoxin A. Sequences which were found to most closely resemble crotoxin B were run in a sequence alignment with crotoxin B from C. d. terrificus, sistruxin B from S. c. tergeminus, and N6 PLA2s from Bothriechis schlegelii and Crotalus viridis viridis. The sequence alignments were made using ClustalW2 from the European Bioinformatics Institute.
Protein Analysis
A protein gel was run using crude venom from both snake species. This gel was run using a 1D 12% polyacrylamide Bis-Tris NuPAGE gel from Life Technologies.The purpose of this gel was to analyze the protein contents of both venoms in order to ensure that they really did contain PLA2 venoms which would be produced by the transcripts isolated in this study.

